Maladie de Creutzfeldt-Jakob : notre action
La surveillance épidémiologique des MCJ menée par Santé publique France a pour principal objectif de suivre la tendance des cas de vMCJ et l’efficacité des mesures mises en place (interdiction des farines niamales dans l’alimentation des bovins). En outre, la surveillance a pour but de détecter le plus précocement l’émergence de nouvelles formes des CMJ et d’en investiguer l’origine et la transmissibilité.
La surveillance épidémiologique de la maladie de Creutzfeldt-Jakob
Le réseau national de surveillance des MCJ et la déclaration obligatoire
Depuis 1992, l’épidémiologie de la maladie de Creutzfeldt-Jakob (MCJ) est sous la surveillance d’un réseau regroupant des laboratoires, des neurologues et des neuropathologistes volontaires, coordonnés par l'Unité 1127 (anciennement U975) de l'Institut national de la santé et de la recherche médicale (Inserm). En outre, les suspicions de MCJ et autres encéphalopathies subaiguës spongiformes transmissibles (ESST) humaines, ont été inscrites à partir du 19 septembre 1996, sur la liste des maladies à déclaration obligatoire (DO). Depuis cette date, une collaboration étroite a été mise en œuvre entre Santé publique France et l’Inserm U1127.
Dans le contexte de l’émergence du variant de la MCJ (vMCJ) et de l’élargissement des sources de données potentielles, en particulier du fait de l’utilisation de la recherche de la protéine 14–3–3 comme critère de suspicion de MCJ, Santé publique France a initié début 2000, avec les différents partenaires concernés, un renforcement de cette surveillance.
En France, 2958 décès liés à une maladie de Creutzfeldt-Jakob certaine ou probable de 1992 à 2017. Parmi les 2958 cas en France : 2543 (86 %) cas sporadiques ; 135 cas iatrogène ; 257 cas génétiques et 27 cas vMCJ.
Les outils de la déclaration obligatoire
La déclaration obligatoire consiste à recueillir des informations aussi exhaustives que possible concernant toutes les suspicions de cas de maladie de Creutzfeldt-Jakob auprès des professionnels. Elle met en jeu deux procédures successives : le signalement et la notification.
Les médecins et les biologistes qui suspectent une maladie de Creutzfeldt-Jakob (MCJ) ou une maladie apparentée doivent en faire le signalement sans délai et par tout moyen approprié (téléphone, télécopie) au médecin de l'Agence régionale de santé (ARS) de leur lieu d'exercice. Dès qu'un signalement de suspicion de MCJ parvient à l'ARS, tous les éléments de ce signalement sont transmis dans les plus brefs délais à Santé publique France. Des suspicions sont également notifiées directement au RNS-MCJ.
Les éléments disponibles au moment de la notification ne permettent pas toujours de classer immédiatement la suspicion en cas possible, probable ou confirmé. En fonction de l’évolution du patient et des résultats des examens (EEG, IRM, etc.), la classification du cas, réalisée à l’aide de la définition de cas (cf. infra), peut prendre plusieurs mois à plusieurs années.
Une attention particulière est apportée à la notion de don de sang par le patient avant la déclaration de la maladie. En effet, en raison de la transmission possible du variant de la MCJ par le sang ou les produits et médicaments dérivés du sang, si un patient suspect de MCJ a récemment été donneur de sang, Santé publique France en lien avec l’EFS, recherche d’éventuels produits et médicaments dérivés du sang toujours disponibles afin de les exclure.
Après le signalement, les médecins ou les biologistes qui suspectent une MCJ doivent en faire la notification au médecin inspecteur de santé publique de l'ARS de leur lieu d'exercice en remplissant une fiche de notification spécifique de suspicion de maladie de Creutzfeldt-Jakob.
Dès que cette fiche parvient à l'ARS, elle est validée, anonymisée et transmise à Santé publique France sans attendre les éléments d'évolution de la maladie ni la confirmation du diagnostic.
Pour déclarer la maladie

Fiche de notification de suspicion de maladie de Creutzfeldt-Jakob

Fiche de procédure en cas de suspicion de MCJ - Pour les cliniciens
Information des personnes sur la notification des maladies à déclaration obligatoire
La définition des cas de maladie de Creutzfeldt-Jakob
La surveillance de la maladie de repose sur des critères de classification des MCJ pour la surveillance des différentes formes : sporadiques, génétiques, acquises ou la forme variante
Formes sporadiques
- Confirmée : Confirmation neuropathologique ou immunocytochimique
- Probable
I + 2 de II + III
ou I + 2 de II + IV (à partir du 1er janvier 2010)
ou MCJ possible + 14-3-3 positive
- Possible : I + 2 de II + durée inférieure à 2 ans
Formes génétiques
- Confirmée
Encéphalopathie subaiguë spongiforme transmissibles (ESST) confirmée à partir du 1er janvier 2010 + ESST définie ou probable chez un apparenté du 1er degré ESST définie avec une mutation pathogène du gène PRNP
- Probable
Trouble neuropsychiatrique progressif + ESST définie ou probable chez un apparenté du 1er degré.
Trouble neuropsychiatrique progressif + mutation pathogène du gène PRNP.
Formes acquises
- Confirmée : ESST confirmée avec un facteur de risque iatrogène reconnu
- Probable
Syndrome cérébelleux prédominant progressif chez un patient traité par hormone hypophysaire
ou ESST probable avec un facteur de risque iatrogène reconnu.
vMCJ
- Cas confirmé : IA et confirmation neuropathologique de v-CJD (e)
- Cas probables
I et 4/5 de II et IIIA et IIIB
ou I et IVA
Facteurs de risque pertinents pour la classification d'un cas de MCJ iatrogène
La pertinence de l’exposition à un facteur de risque doit tenir compte du délai entre l’exposition et le début de la maladie :
- Traitement par hormone de croissance hypophysaire humaine, hormone gonadotrope hypophysaire humaine ou dure mère humaine ;
- Greffe de cornée si le donneur de la cornée était atteint d’une maladie à prion définie ou probable ;
- Exposition à des instruments de neurochirurgie utilisés au préalable chez un patient atteint de maladie à prion humaine définie ou probable.
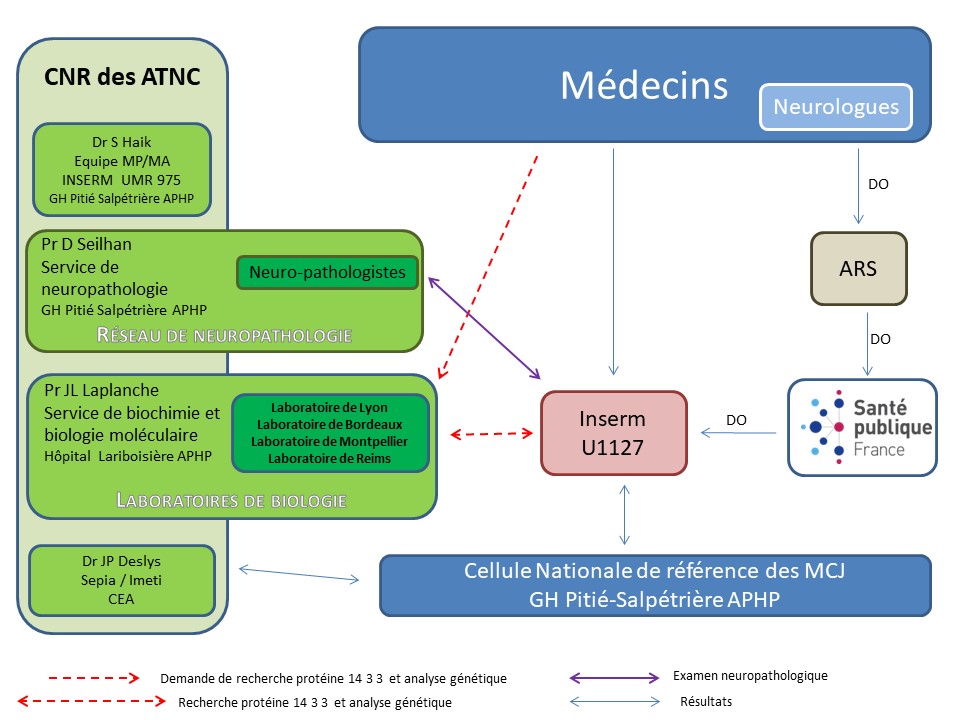
Le réseau national de surveillance des maladies de Creutzfeldt-Jakob et maladies apparentées.
Depuis l’inscription des suspicions de MCJ et autres encéphalopathies subaiguës spongiformes transmissibles (ESST) humaines sur la liste des maladies à déclaration obligatoire, le 19 septembre 1996, une collaboration étroite a vu le jour entre Santé publique France et l’Inserm U1127.
Santé publique France a initié début 2000, avec les différents partenaires concernés, un renforcement de cette surveillance à travers la création du Réseau national de surveillance des maladies de Creutzfeldt-Jakob et des maladies apparentées.
Ce réseau est constitué de différents acteurs :
- Neurologues,
- Unité 975 de l’Inserm,
- Centre national de référence (CNR) des agents transmissibles non conventionnels (ATNC), composé de trois laboratoires : le service d'anatomie pathologique-neurologie du Centre hospitalier Pitié-Salpêtrière, Assistance Publique-Hôpitaux de Paris qui coordonne le Réseau de neuropathologie pour la MCJ composé de 22 centres régionaux de neuropathologie ; le service de biochimie et de biologie moléculaire de l'hôpital Lariboisière (Assistance Publique-Hôpitaux de Paris), qui anime un réseau de laboratoires ; et l'équipe Inserm Avenir – Maladies à prions chez l’homme, IFR des neurosciences, Groupe hospitalier Pitié-Salpêtrière,
- La Cellule nationale de référence des MCJ
- Les Agence régionales de santé,
- Santé publique France.
Coordonné par Santé publique France, ce réseau, est formalisé par une convention tri-annuelle entre Santé publique France et l’Inserm. Ses objectifs sont de :
- Détecter tous les cas d’encéphalopathie subaiguë spongiforme transmissibles (ESST) humaines, et plus particulièrement les cas de v-MCJ,
- Les classer par étiologie,
- En estimer l'incidence,
- Décrire leur répartition temporo-spatiale, leurs tendances évolutives,
- Détecter des cas groupés.